Research
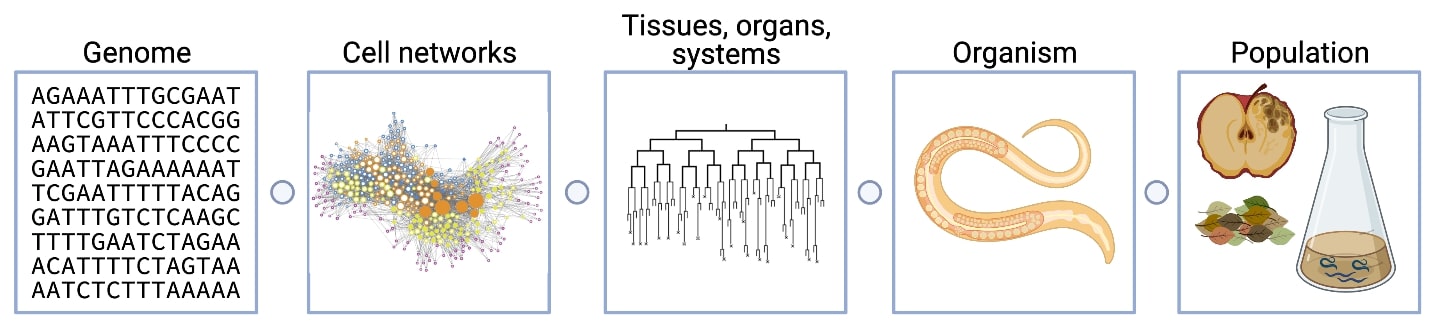
Model organisms provide the opportunity to experimentally test the correlations between genetic variation and trait differences across populations because of the ease of manipulation and powerful tools. However, most model organism research is based on a single wild-type strain background with little connection to natural variation, which is like studying a single person to make conclusions about the entire human species. Selfing Caenorhabditis species, including C. elegans, are isolated worldwide, have ample variation, rich ecological histories, and experimentally tractable traits and genomes. Therefore, these species provide the opportunities to identify the genes that vary among individuals and the molecular mechanisms for how genetic variation causes phenotypic differences. We can use variation within and across different Caenorhabditis species. Our lab uses a variety of genetic and genomic tools to discover the molecular mechanisms of evolutionary change in Caenorhabditis natural populations. The range of our research projects from genomes to organisms to ecology is shown above. Some of our ongoing projects are described below.
- The molecular mechanisms of gene-by-environment interactions
- Genome evolution of selfing Caenorhabditis species
- Ecological sampling of wild Caenorhabditis species
- Life-history trait evolution
- Genetic causes of resistance to anthelmintic compounds
- Evolution of cellular networks
The molecular mechanisms of gene-by-environment interactions
To map differences in quantitative traits, we use high-throughput assays to rapidly measure gene-by-environment interactions across large collections of natural C. elegans, C. briggsae, and C. tropicalis strains. These assays measure fecundity, growth rates, behaviors, and pharyngeal pumping rates after exposure to chemotherapeutics, toxicants, and other compounds. Each trait allows us to determine different effects on the physiology of the animals. These high-throughput assays give us the ability to identify natural variants or induced mutations at unprecedented speeds and with high statistical power. To enable the connections between genotypic and phenotypic variation, we created a new genome-wide association mapping package called NemaScan.
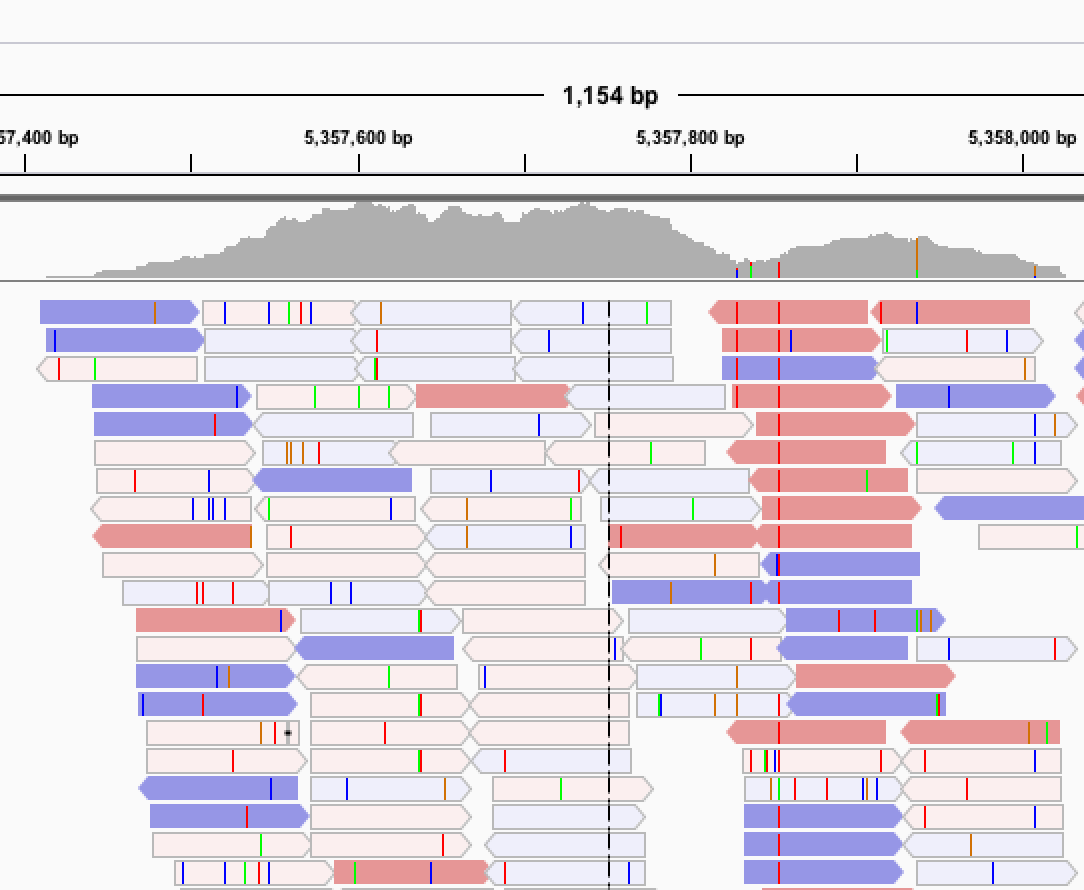
Genome evolution of selfing Caenorhabditis species
Because of the efforts of a number of highly dedicated scientists and citizen volunteers, we have amassed a large collection of C. elegans, C. briggsae, and C. tropicalis wild strains from throughout the world. These strains represent nearly all known isolation locations where we have found these species. Our goals are to use this large strain collection to identify functional variation for many quantitative traits and to understand more about the population genetic forces shaping these genomes. As a first step, we are creating new telomere-to-telomere reference genomes and manually curated de novo gene models.
Next, we are actively deep sequencing all of the strains for each of the three species using Illumina and PacBio platforms. These large sequence data sets allow us to not only identify single nucleotide variants but to dig deeper into other classes of genetic variation, including insertion/deletions, transposon insertions, genomic rearrangements, short-tandem repeats, and copy number variants. We hope that these additional classes of variation will contribute to our understanding of how genetic variation underlies phenotypic variation and genome evolution.

Ecological sampling of wild Caenorhabditis species
To discover additional diversity in the three selfing species, C. elegans, C. briggsae, and C. tropicalis, and to identify new Caenorhabditis species, we sample wild nematodes from throughout the world. Our most recent focus is on natural diversity from the Hawaiian Islands, where we have found the most genetically diverse strains for all three species. These species are enriched in rotting fruits, flowers, and vegetal matter across a range of elevations and niches. Our longitudinal collections span multiple years, so we have cryopreserved and whole-genome sequenced strains from numerous habitats that can be connected to evolution of traits specific to Hawaiian niches. We continue to expand sampling and data collection from this field work to include microbiome sampling, measurements of ethanol content, metals, carbon content, and other environmental parameters.
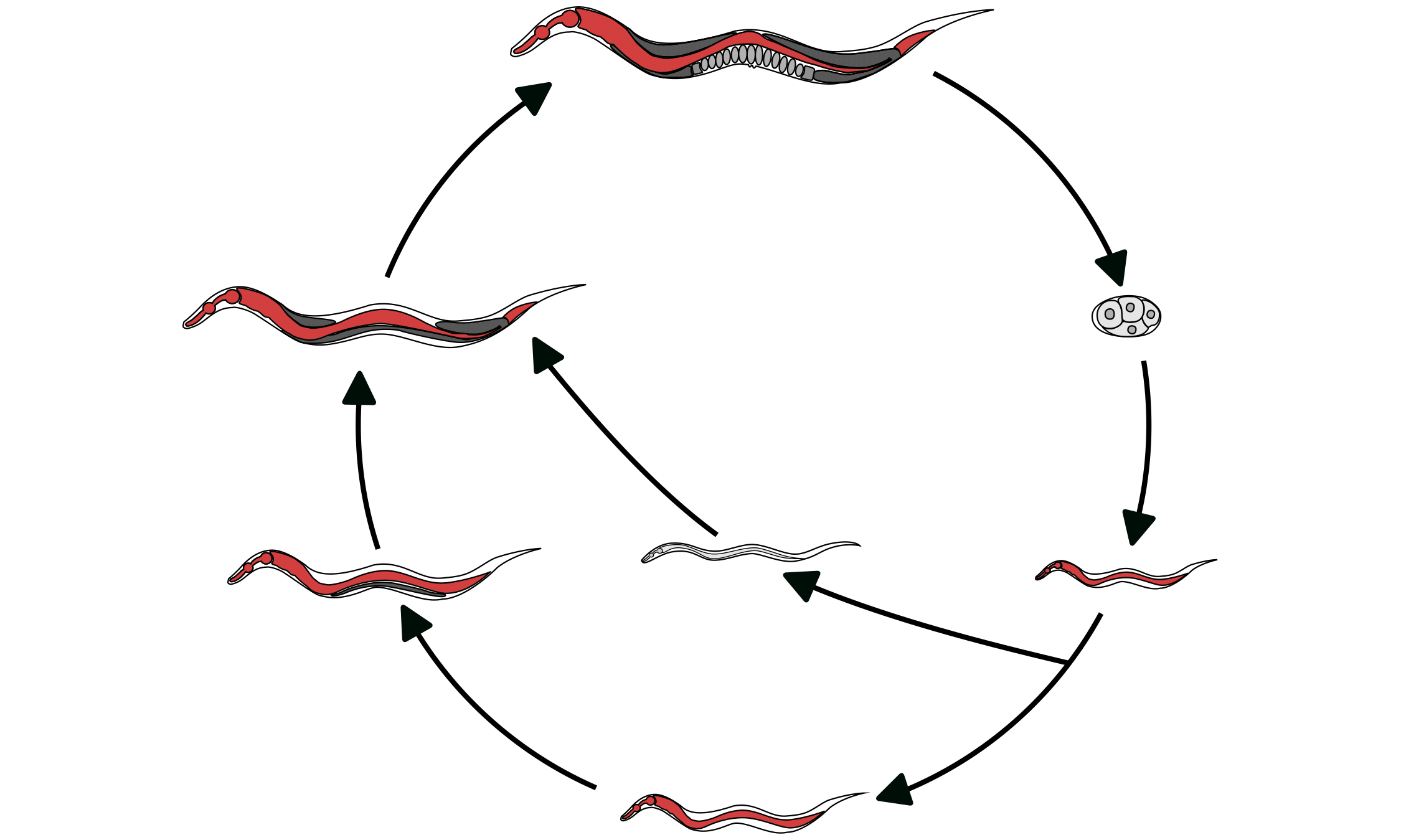
Life-history trait evolution
Organisms must adapt to changing environments like temperature and food availability. Caenorhabditis nematodes enter an alternative developmental stage when poor conditions are encountered. Using novel high-throughput assays, we found that natural populations vary in their propensity to enter this alternative developmental stage. Additionally, in collaboration with the Schroeder lab (Cornell Univ.), we found that nematodes produce varying amounts of the chemical cues that induce the alternative developmental stage. Taken together, we hope to understand how nematodes adapt to environmental cues to alter developmental trajectories.
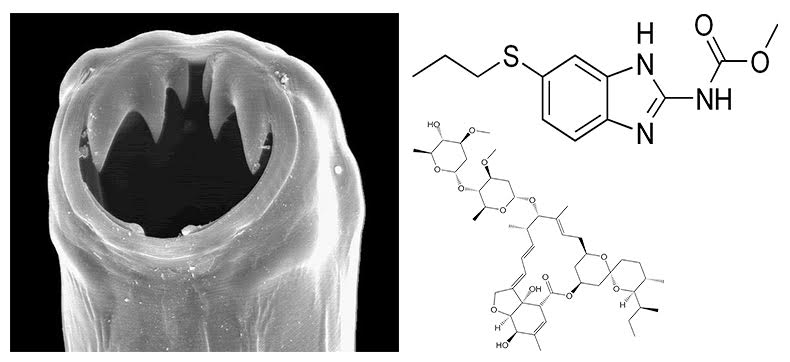
Genetic causes of resistance to anthelmintic compounds
Using high-throughput assays that measure organismal fitness (offspring production, growth rate) and behaviors (feeding rate and paralysis), we are investigating how three clade V nematode species, C. briggsae, C. elegans, and C. tropicalis vary in responses to anthelmintic (anti-nematode) compounds. Our goal is to identify resistance mechanisms conserved with parasitic roundworms to better treat infected people in developing countries. Our recent focus is on the genetic variants that mediate macrocyclic lactone (ML) and benzimidazole (BZ) resistance across natural populations. For MLs, we have identified at least five other loci that control resistance in natural populations beyond variation in the glutamate-gated chloride channel gene, glc-1. For BZs, we found a new genomic locus involved in resistance and also high levels of natural heterogeneity in the benzimidazole target ben-1. We are collaborating with parasite research groups to validate our Caenorhabditis findings in tractable parasitic helminths. Extending beyond Caenorhabditis, we have started quantitative genetics and new genomes for the turkey ascarid Ascaridia dissimilis. We believe that these projects will elucidate how ascarids become resistant to BZ drugs because the mechanisms seem to be independent from the standard beta-tubulin target gene.
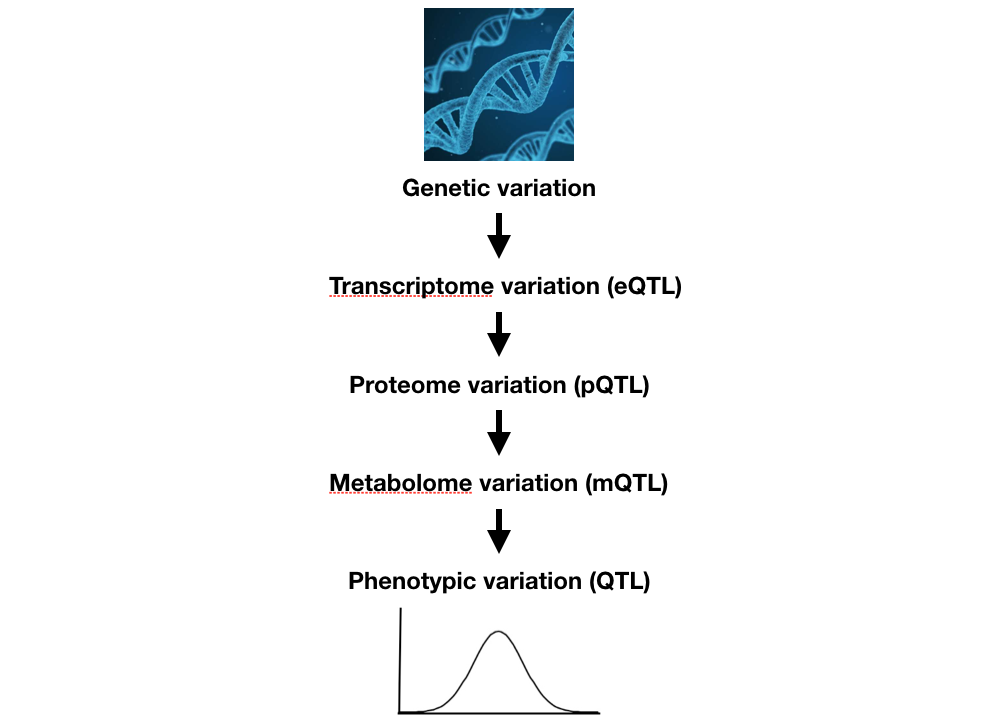
Evolution of cellular networks
In addition to toxicant responses, anthelmintic resistance, behaviors, and development, we are measuring the abundances of mRNAs, small RNAs, proteins, and metabolites across hundreds of natural C. elegans, C. briggsae, and C. tropicalis isolates. We hope to use these data to better refine how organismal traits vary across populations and investigate evolutionary trends. Additionally, we have shown that these intermediate traits are statistical mediators that help us to more rapidly identify causal genetic loci underlying quantitative trait differences.