nil-ril-nf¶
The nil-ril-nf pipeline will align, call variants, and generate datasets for NIL and RIL sequence data. It runs a hidden-markov-model to fill in missing genotypes from low-coverage sequence data.
Docker image¶
The docker image used by the nil-ril-nf pipeline is the nil-ril-nf docker image:
andersenlab/nil-ril-nf¶
The Dockerfile is stored in the root of the nil-nf github repo and is automatically built on Dockerhub whenever the repo is pushed.
Usage¶
███╗ ██╗██╗██╗ ██████╗ ██╗██╗ ███╗ ██╗███████╗
████╗ ██║██║██║ ██╔══██╗██║██║ ████╗ ██║██╔════╝
██╔██╗ ██║██║██║█████╗██████╔╝██║██║█████╗██╔██╗ ██║█████╗
██║╚██╗██║██║██║╚════╝██╔══██╗██║██║╚════╝██║╚██╗██║██╔══╝
██║ ╚████║██║███████╗ ██║ ██║██║███████╗ ██║ ╚████║██║
╚═╝ ╚═══╝╚═╝╚══════╝ ╚═╝ ╚═╝╚═╝╚══════╝ ╚═╝ ╚═══╝╚═╝
parameters description Set/Default
========== =========== =======
--debug Set to 'true' to test false
--cores Number of cores 4
--A Parent A N2
--B Parent B CB4856
--cA Parent A color (for plots) #0080FF
--cB Parent B color (for plots) #FF8000
--out Directory to output results NIL-N2-CB4856-2017-09-27
--fqs fastq file (see help) (required)
--relative use relative fastq prefix ${params.relative}
--reference Reference Genome /Users/dancook/Documents/git/nil-nf/reference/WS245.fa.gz
--vcf VCF to fetch parents from (required)
--tmpdir A temporary directory tmp/
The Set/Default column shows what the value is currently set to
or would be set to if it is not specified (it's default).
Overview¶
The nil-ril-nf pipeline:

Alignment- Performed using bwa-memMerge Bams- Combines bam files aligned individually for each fastq-pair. Sambamba is actually used in place of samtools, but it's a drop-in, faster replacement.Bam Stats- A variety of metrics are calculated for bams and combined into individual files for downstream analsyis.Mark Duplicates- Duplicate reads are marked using Picard.Call Variants individual- Variants are called for each strain inidividually first. This generates a sitelist which is used to identify all variant sites in the population.Pull parental genotypes- Pulls out parental genotypes from the given VCF. The list of genotypes is filtered for discordant calls (i.e. different genotypes). This is VCF is used to generate a sitelist for calling low-coverage bams and later is merged into the resulting VCF.Call variants union- Uses the sitelist from the previous step to call variants on low-coverage sequence data. The resulting VCF will have a lot of missing calls.Merge VCF- Merges in the parental VCF (which has been filtered only for variants with discordant calls).Call HMM- VCF-kit is run in various ways to infer the appropriate genotypes from the low-coverage sequence data.
Docker image¶
The docker image used by the nil-ril-nf pipeline is the nil-ril-nf docker image:
The Dockerfile is stored in the root of the nil-ril-nf github repo and is automatically built on Dockerhub whenever the repo is pushed.
Usage¶
Profiles and Running the Pipeline¶
The nextflow.config file included with this pipeline contains four profiles. These set up the environment for testing local development, testing on Quest, and running the pipeline on Quest.
local- Used for local development. Uses the docker container.quest_debug- Runs a small subset of available test data. Should complete within a couple of hours. For testing/diagnosing issues on Quest.quest- Runs the entire dataset.travis- Used by travis-ci for testing purposes.
Running the pipeline locally¶
When running locally, the pipeline will run using the andersenlab/nil-ril-nf docker image. You must have docker installed. You will need to obtain a reference genome to run the alignment with as well. You can use the following command to obtain the reference:
curl https://storage.googleapis.com/elegansvariation.org/genome/WS245/WS245.tar.gz > WS245.tar.gz
tar -xvzf WS245.tar.gz
Run the pipeline locally with:
nextflow run main.nf -profile local -resume
Debugging the pipeline on Quest¶
When running on Quest, you should first run the quest debug profile. The Quest debug profile will use a test dataset and sample sheet which runs much faster and will encounter errors much sooner should they need to be fixed. If the debug dataset runs to completion it is likely that the full dataset will as well.
nextflow run main.nf -profile quest_debug -resume
Running the pipeline on Quest¶
The pipeline can be run on Quest using the following command:
nextflow run main.nf -profile quest -resume
Testing¶
If you are going to modify the pipeline, I highly recommend doing so in a testing environment. The pipeline includes a debug dataset that runs rather quickly (~10 minutes). If you cache results initially and re-run with the -resume option it is fairly easy to add new processes or modify existing ones and still ensure that things are output correctly.
Additionally - note that the pipeline is tested everytime a change is made and pushed to github. Testing takes place on travis-ci here, and a badge is visible on the readme indicating the current 'build status'. If the pipeline encounters any errors when being run on travis-ci the 'build' will fail.
The command below can be used to test the pipeline locally.
# Downloads a pre-indexed reference
curl https://storage.googleapis.com/elegansvariation.org/genome/WS245/WS245.tar.gz > WS245.tar.gz
tar -xvzf WS245.tar.gz
# Run nextflow
nextflow run andersenlab/nil-ril-nf \
-with-docker andersenlab/nil-ril-nf \
--debug \
--reference=WS245.fa.gz \
-resume
- Note that the path to the vcf will change slightly in releases later than WI-20170531; See the
wi-nfpipeline for details. - The command above will automatically place results in a folder:
NIL-N2-CB4856-YYYY-MM-DD
Parameters¶
--debug¶
The pipeline comes pre-packed with fastq's and a VCF that can be used to debug. See the Testing section for more information.
--cores¶
The number of cores to use during alignments and variant calling.
--A, --B¶
Two parental strains must be provided. By default these are N2 and CB4856. The parental strains provided must be present in the VCF provided. Their genotypes are pulled from that VCF and used to generate the HMM. See below for more details.
--cA, --cB¶
The color to use for parental strain A and B on plots.
--out¶
A directory in which to output results. By default it will be NIL-A-B-YYYY-MM-DD where A and be are the parental strains.
--fqs (FASTQs)¶
In order to process NIL/RIL data, you need to move the sequence data to a folder and create a fq_sheet.tsv. This file defines the fastqs that should be processed. The fastq can be specified as relative or absolute. By default, they are expected to be relative to the fastq file. The FASTQ sheet details strain names, ids, library, and files. It should be tab-delimited and look like this:
NIL_01 NIL_01_ID S16 NIL_01_1.fq.gz NIL_01_2.fq.gz
NIL_02 NIL_02_ID S1 NIL_02_1.fq.gz NIL_02_2.fq.gz
Notice that the file does not include a header. The table with corresponding columns looks like this.
| strain | fastq_pair_id | library | fastq-1-path | fastq-2-path |
|---|---|---|---|---|
| NIL_01 | NIL_01_ID | S16 | NIL_01_1.fq.gz | NIL_01_2.fq.gz |
| NIL_02 | NIL_02_ID | S1 | NIL_02_1.fq.gz | NIL_02_2.fq.gz |
The columns are detailed below:
- strain - The name of the strain. If a strain was sequenced multiple times this file is used to identify that fact and merge those fastq-pairs together following alignment.
- fastq_pair_id - This must be unique identifier for all individual FASTQ pairs.
- library - A string identifying the DNA library. If you sequenced a strain from different library preps it can be beneficial when calling variants. The string can be arbitrary (e.g. LIB1) as well if only one library prep was used.
- fastq-1-path - The relative path of the first fastq.
- fastq-2-path - The relative path of the second fastq.
This file needs to be placed along with the sequence data into a folder. The tree will look like this:
NIL_SEQ_DATA/
├── NIL_01_1.fq.gz
├── NIL_01_2.fq.gz
├── NIL_02_1.fq.gz
├── NIL_02_2.fq.gz
└── fq_sheet.tsv
Set --fqs as --fqs=/the/path/to/fq_sheet.tsv.
Important
Do not perform any pre-processing on NIL data. NIL-data is low-coverage by design and you want to retain as much sequence data (however poor) as possible.
If you want to specify fastqs using an absolute path use --relative=false
--relative¶
Set to true by default. If you set --relative=false, fq's in the fq_sheet are expected to use an absolute path.
--vcf (Parental VCF)¶
Before you begin, you will need access to a VCF with high-coverage data from the parental strains. In general, this can be obtained using the latest release of the wild-isolate data which is usually located in the b1059 analysis folder. For example, you would likely want to use:
/projects/b1059/analysis/WI-20170531/vcf/WI.20170531.hard-filter.vcf.gz
This is the hard-filtered VCF, meaning that poor quality variants have been stripped. Use hard-filtered VCFs for this pipeline.
Set the parental VCF as --vcf=/the/path/to/WI.20170531.hard-filter.vcf.gz
--reference¶
A fasta reference indexed with BWA. On Quest, the reference is available here:
/projects/b1059/data/genomes/c_elegans/WS245/WS245.fa.gz
--tmpdir¶
A directory for storing temporary data.
Output¶
The final output directory looks like this:
.
├── log.txt
├── fq
│ ├── fq_bam_idxstats.tsv
│ ├── fq_bam_stats.tsv
│ ├── fq_coverage.full.tsv
│ └── fq_coverage.tsv
├── SM
│ ├── SM_bam_idxstats.tsv
│ ├── SM_bam_stats.tsv
│ ├── SM_coverage.full.tsv
│ ├── SM_union_vcfs.txt
│ └── SM_coverage.tsv
├── hmm
│ ├── gt_hmm.(png/svg)
│ ├── gt_hmm.tsv
│ ├── gt_hmm_fill.tsv
│ ├── NIL.filtered.stats.txt
│ ├── NIL.filtered.vcf.gz
│ ├── NIL.filtered.vcf.gz.csi
│ ├── NIL.hmm.vcf.gz
│ ├── NIL.hmm.vcf.gz.csi
│ └── gt_hmm_genotypes.tsv
├── bam
│ └── <BAMS + indices>
├── duplicates
│ └── bam_duplicates.tsv
└─ sitelist
├── N2.CB4856.sitelist.[tsv/vcf].gz
└── N2.CB4856.sitelist.[tsv/vcf].gz.[tbi/csi]
log.txt¶
A summary of the nextflow run.
duplicates/¶
bam_duplicates.tsv - A summary of duplicate reads from aligned bams.
fq/¶
- fq_bam_idxstats.tsv - A summary of mapped and unmapped reads by fastq pair.
- fq_bam_stats.tsv - BAM summary by fastq pair.
- fq_coverage.full.tsv - Coverage summary by chromosome
- fq_coverage.tsv - Simple coverage file by fastq
SM/¶
If you have multiple fastq pairs per sample, their alignments will be combined into a strain or sample-level BAM and the results will be output to this directory.
- SM_bam_idxstats.tsv - A summary of mapped and unmapped reads by sample.
- SM_bam_stats.tsv - BAM summary at the sample level
- SM_coverage.full.tsv - Coverage at the sample level
- SM_coverage.tsv - Simple coverage at the sample level.
- SM_union_vcfs.txt - A list of VCFs that were merged to generate RIL.filter.vcf.gz
hmm/¶
Important
gt_hmm_fill.tsv is for visualization purposes only. To determine breakpoints you should use gt_hmm.tsv.
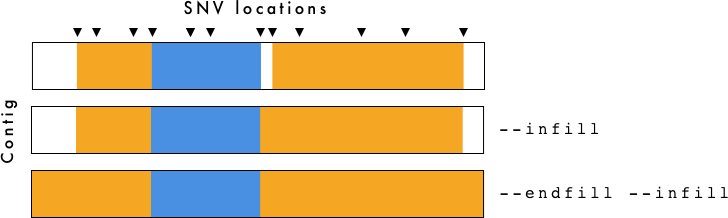
The --infill and --endfill options are applied to the gt_hmm_fill.tsv file. You need to be cautious when examining this data as it is generated primarily for visualization purposes.
- gt_hmm.(png/svg) - Haplotype plot using
--infilland--endfill. - gt_hmm_fill.tsv - Same as above, but using
--infilland--endfillwith VCF-Kit. For more information, see VCF-Kit Documentation. This file is used to generate the plots. - gt_hmm.tsv - Haplotypes defined by region with associated information. Does not use
--infilland--endfill - gt_hmm_genotypes.tsv - Long form genotypes file.
- NIL/RIL.filtered.vcf.gz - A VCF genotypes including the NILs and parental genotypes.
- NIL/RIL.filtered.stats.txt - Summary of filtered genotypes. Generated by
bcftools stats NIL.filtered.vcf.gz - NIL/RIL.hmm.vcf.gz - The NIL/RIL VCF as output by VCF-Kit; HMM applied to determine genotypes.
plots/¶
- coverage_comparison.(png/svg/pdf) - Compares FASTQ and Sample-level coverage. Note that coverage is not simply cumulative. Only uniquely mapped reads count towards coverage, so it is possible that the sample-level coverage will not equal to the cumulative sum of the coverages of individual FASTQ pairs.
- duplicates.(png/svg/pdf) - Coverage vs. percent duplicated.
- unmapped_reads.(png/svg/pdf) - Coverage vs. unmapped read percent.
sitelist/¶
<A>.<B>.sitelist.tsv.gz[+.tbi]- A tabix-indexed list of sites found to be different between both parental strains.<A>.<B>.sitelist.vcf.gz[+.tbi]- A vcf of sites found to be different between both parental strains.