wi-nf¶
The wi-nf pipeline aligns, calls variants, and performs analysis from wild isolate sequence data. The output of the wi-nf pipeline can be uploaded to google storage as a new release for the CeNDR website.
Usage¶
▄ ▄ ▄▄▄▄▄▄▄▄▄▄▄ ▄▄ ▄ ▄▄▄▄▄▄▄▄▄▄▄
▐░▌ ▐░▌▐░░░░░░░░░░░▌ ▐░░▌ ▐░▌▐░░░░░░░░░░░▌
▐░▌ ▐░▌ ▀▀▀▀█░█▀▀▀▀ ▐░▌░▌ ▐░▌▐░█▀▀▀▀▀▀▀▀▀
▐░▌ ▐░▌ ▐░▌ ▐░▌▐░▌ ▐░▌▐░▌
▐░▌ ▄ ▐░▌ ▐░▌ ▄▄▄▄▄▄▄▄▄▄▄ ▐░▌ ▐░▌ ▐░▌▐░█▄▄▄▄▄▄▄▄▄
▐░▌ ▐░▌ ▐░▌ ▐░▌ ▐░░░░░░░░░░░▌ ▐░▌ ▐░▌ ▐░▌▐░░░░░░░░░░░▌
▐░▌ ▐░▌░▌ ▐░▌ ▐░▌ ▀▀▀▀▀▀▀▀▀▀▀ ▐░▌ ▐░▌ ▐░▌▐░█▀▀▀▀▀▀▀▀▀
▐░▌▐░▌ ▐░▌▐░▌ ▐░▌ ▐░▌ ▐░▌▐░▌▐░▌
▐░▌░▌ ▐░▐░▌ ▄▄▄▄█░█▄▄▄▄ ▐░▌ ▐░▐░▌▐░▌
▐░░▌ ▐░░▌▐░░░░░░░░░░░▌ ▐░▌ ▐░░▌▐░▌
▀▀ ▀▀ ▀▀▀▀▀▀▀▀▀▀▀ ▀ ▀▀ ▀
parameters description Set/Default
========== =========== =======
--debug Set to 'true' to test ${params.debug}
--cores Regular job cores ${params.cores}
--out Directory to output results ${params.out}
--fqs fastq file (see help) ${params.fqs}
--fq_file_prefix fastq prefix ${params.fq_file_prefix}
--reference Reference Genome ${params.reference}
--annotation_reference SnpEff annotation ${params.annotation_reference}
--bamdir Location for bams ${params.bamdir}
--tmpdir A temporary directory ${params.tmpdir}
--email Email to be sent results ${params.email}
HELP: http://andersenlab.org/dry-guide/pipeline-wi/
Pipeline Overview¶
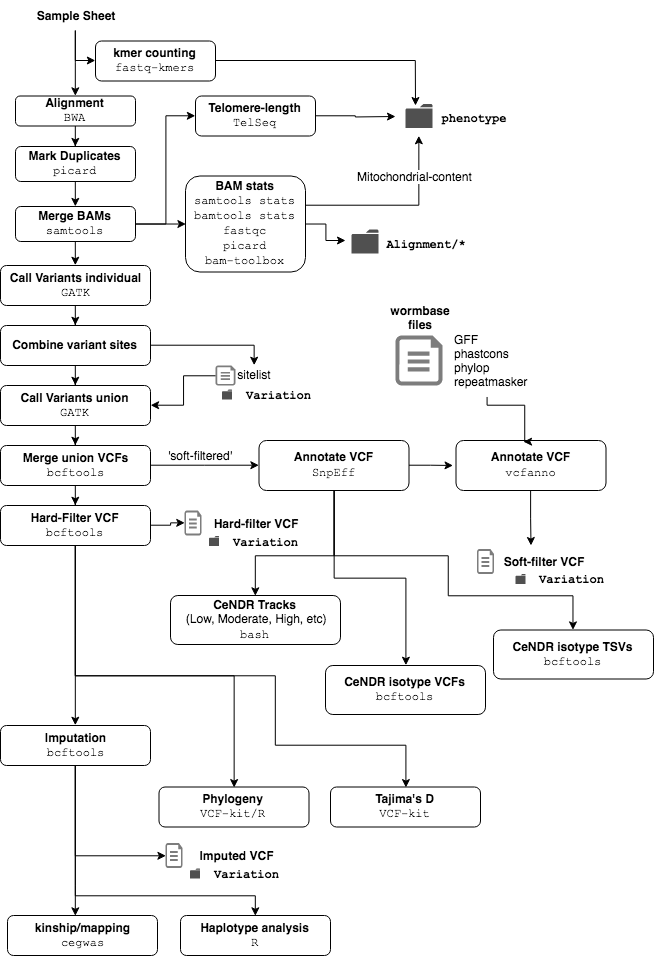
Usage¶
Docker File¶
andersenlab/wi-nf is the docker image used within the wi-nf pipeline. If Quest ever supports singularity, it can be converted to a singularity image and used with Nextflow.
pyenv environments¶
The pipeline uses two python environments due to clashing dependencies. The two environments are:
vcf-kit- A python 2.7.14 environment with vcf-kit and cyvcf installed.multiqc- A python 3.6.0 environment with multiqc installed.
If you are not using the docker container you must install these virtual environments by running the setup_pyenv.sh script:
bash setup_pyenv.sh
If you require one of these environments for a process within the pipeline, add the following as the first line of your script:
source init_pyenv.sh && pyenv activate <environment name>
Profiles and Running the Pipeline¶
The nextflow.config file included with this pipeline contains three profiles. These set up the environment for testing local development, testing on Quest, and running the pipeline on Quest.
local- Used for local development. Uses the docker container.quest_debug- Runs a small subset of available test data. Should complete within a couple of hours. For testing/diagnosing issues on Quest.quest- Runs the entire dataset.
Running the pipeline locally¶
When running locally, the pipeline will run using the andersenlab/wi-nf docker image. You must have docker installed.
nextflow run main.nf -profile local -resume
Debugging the pipeline on Quest¶
When running on Quest, you should first run the quest debug profile. The Quest debug profile will use a test dataset and sample sheet which runs much faster and will encounter errors much sooner should they need to be fixed. If the debug dataset runs to completion it is likely that the full dataset will as well.
nextflow run main.nf -profile quest_debug -resume
Running the pipeline on Quest¶
The pipeline can be run on Quest using the following command:
nextflow run main.nf -profile quest -resume
Configuration¶
Most configuration is handled using the -profile flag and nextflow.config; If you want to fine tune things you can use the options below.
--cores¶
The number of cores to use during alignments and variant calling.
--out¶
A directory in which to output results. By default it will be WI-YYYY-MM-DD where YYYY-MM-DD is todays date.
--fqs (FASTQs)¶
When running the wi-nf pipeline you must provide a sample sheet that tells it where fastqs are and which samples group into isotypes. By default, this is the sample sheet in the base of the wi-nf repo (SM_sample_sheet.tsv), but can be specified using --fqs if an alternative is required. The sample sheet provides information on the isotype, fastq_pairs, library, location of fastqs, and sequencing folder.
More information on the sample sheet and adding new sequence data in the Sample sheet section.
--fqs_file_prefix¶
A prefix path for FASTQs defined in a sample sheet. The sample sheet designed for usage on Quest (SM_sample_sheet.tsv) uses absolute paths so no FASTQ prefix is necessary. Additionally, there is no need to set this option as it is set for you when using the -profile flag. This option is only necessary (maybe) with a custom dataset where you are not using absolute paths to reference FASTQs.
--reference¶
A fasta reference indexed with BWA. On Quest, the reference is available here:
/projects/b1059/data/genomes/c_elegans/WS245/WS245.fa.gz
--tmpdir¶
A directory for storing temporary data.
Sample Sheet¶
The sample sheet defines which FASTQs belong to which isotypes, set FASTQ IDs, library, and more. The sample sheet is constructed using the scripts/construct_SM_sheet.sh script. When new sequence data is added, this needs to be modified to add information about the new FASTQs. Updates to this script should be committed to the repo.
Important
You have to assign isotypes using the concordance script before they can be processed using wi-nf.
When you run the scripts/construct_SM_sheet.sh, it will output the SM_Sample_sheet.tsv file in the base of the repo. You should carefully examine the diff of this file using Git to ensure it is modifying the sample sheet correctly. Errors can be disasterous. Note that the output uses absolute paths to FASTQ files.
Sample sheet structure
AB1 BGI1-RET2-AB1 RET2 /projects/b1059/data/fastq/WI/dna/processed/original_wi_set/BGI1-RET2-AB1-trim-1P.fq.gz /projects/b1059/data/fastq/WI/dna/processed/original_wi_set/BGI1-RET2-AB1-trim-2P.fq.gz original_wi_set
AB1 BGI2-RET2-AB1 RET2 /projects/b1059/data/fastq/WI/dna/processed/original_wi_set/BGI2-RET2-AB1-trim-1P.fq.gz /projects/b1059/data/fastq/WI/dna/processed/original_wi_set/BGI2-RET2-AB1-trim-2P.fq.gz original_wi_set
AB1 BGI3-RET2b-AB1 RET2b /projects/b1059/data/fastq/WI/dna/processed/original_wi_set/BGI3-RET2b-AB1-trim-1P.fq.gz /projects/b1059/data/fastq/WI/dna/processed/original_wi_set/BGI3-RET2b-AB1-trim-2P.fq.gz original_wi_set
Notice that the file does not include a header. The table with corresponding columns looks like this.
| isotype | fastq_pair_id | library | fastq-1-path | fastq-2-path | sequencing_folder |
|---|---|---|---|---|---|
| AB1 | BGI1-RET2-AB1 | RET2 | /projects/b1059/data/fastq/WI/dna/processed/original_wi_set/BGI1-RET2-AB1-trim-1P.fq.gz | /projects/b1059/data/fastq/WI/dna/processed/original_wi_set/BGI1-RET2-AB1-trim-2P.fq.gz | original_wi_set |
| AB1 | BGI2-RET2-AB1 | RET2 | /projects/b1059/data/fastq/WI/dna/processed/original_wi_set/BGI2-RET2-AB1-trim-1P.fq.gz | /projects/b1059/data/fastq/WI/dna/processed/original_wi_set/BGI2-RET2-AB1-trim-2P.fq.gz | original_wi_set |
| AB1 | BGI3-RET2b-AB1 | RET2b | /projects/b1059/data/fastq/WI/dna/processed/original_wi_set/BGI3-RET2b-AB1-trim-1P.fq.gz | /projects/b1059/data/fastq/WI/dna/processed/original_wi_set/BGI3-RET2b-AB1-trim-2P.fq.gz | original_wi_set |
The columns are detailed below:
- isotype - The name of the isotype. It is used to group FASTQ-pairs into BAMs which are treated as individuals.
- fastq_pair_id - This must be unique identifier for all individual FASTQ pairs. There is (unfortunately) no standard for this.
- library - A name identifying the DNA library. If the FASTQ-pairs for a strain were sequenced using different library preps they should be assigned different library names. Likewise, if they were the same DNA library they should have the same library name. Keep in mind that within an isotype the library names for each strain must be independent.
- fastq-1-path - The absolute path of the first fastq.
- fastq-2-path - The absolute path of the second fastq.
Output¶
The output from the pipeline is structured to enable it to easily be integrated with CeNDR. The final output directory looks like this:
├── log.txt
├── alignment
│ ├── isotype_bam_idxstats.tsv
│ ├── isotype_bam_stats.tsv
│ ├── isotype_coverage.full.tsv
│ └── isotype_coverage.tsv
├── cegwas
│ ├── kinship.Rda
│ └── snps.Rda
├── isotype
│ ├── tsv
│ │ ├── <isotype>.(date).tsv.gz
│ │ └── <isotype>.(date).tsv.gz
│ └── vcf
│ ├── <isotype>.(date).vcf.gz
│ └── <isotype>.(date).vcf.gz.tbi
├── phenotype
│ ├── MT_content.tsv
│ ├── kmers.tsv
│ └── telseq.tsv
├── popgen
│ ├── WI.(date).tajima.bed.gz
│ ├── WI.(date).tajima.bed.gz.tbi
│ └── trees
│ ├── I.pdf
│ ├── I.png
│ ├── I.tree
│ ├── ...
│ ├── genome.pdf
│ ├── genome.png
│ └── genome.tree
├── report
│ ├── multiqc.html
│ └── multiqc_data
│ ├── multiqc_bcftools_stats.json
│ ├── multiqc_data.json
│ ├── multiqc_fastqc.json
│ ├── multiqc_general_stats.json
│ ├── multiqc_picard_AlignmentSummaryMetrics.json
│ ├── multiqc_picard_dups.json
│ ├── multiqc_picard_insertSize.json
│ ├── multiqc_samtools_idxstats.json
│ ├── multiqc_samtools_stats.json
│ ├── multiqc_snpeff.json
│ └── multiqc_sources.json
├── tracks
│ ├── (date).HIGH.bed.gz
│ ├── (date).HIGH.bed.gz.tbi
│ ├── (date).LOW.bed.gz
│ ├── (date).LOW.bed.gz.tbi
│ ├── (date).MODERATE.bed.gz
│ ├── (date).MODERATE.bed.gz.tbi
│ ├── (date).MODIFIER.bed.gz
│ ├── (date).MODIFIER.bed.gz.tbi
│ ├── phastcons.bed.gz
│ ├── phastcons.bed.gz.tbi
│ ├── phylop.bed.gz
│ └── phylop.bed.gz.tbi
└── variation
├── WI.(date).soft-filter.vcf.gz
├── WI.(date).soft-filter.vcf.gz.csi
├── WI.(date).soft-filter.vcf.gz.tbi
├── WI.(date).soft-filter.stats.txt
├── WI.(date).hard-filter.vcf.gz
├── WI.(date).hard-filter.vcf.gz.csi
├── WI.(date).hard-filter.vcf.gz.tbi
├── WI.(date).hard-filter.stats.txt
├── WI.(date).hard-filter.genotypes.tsv
├── WI.(date).hard-filter.genotypes.frequency.tsv
├── WI.(date).impute.vcf.gz
├── WI.(date).impute.vcf.gz.csi
├── WI.(date).impute.vcf.gz.tbi
├── WI.(date).impute.stats.txt
├── sitelist.tsv.gz
└── sitelist.tsv.gz.tbi
log.txt¶
A summary of the nextflow run.
alignment/¶
Alignment statistics by isotype
- isotype_bam_idxstats.tsv - A summary of mapped and unmapped reads by sample.
- isotype_bam_stats.tsv - BAM summary at the sample level
- isotype_coverage.full.tsv - Coverage at the sample level
- isotype_coverage.tsv - Simple coverage at the sample level.
cegwas/¶
- kinship.Rda - A kinship matrix constructed from
WI.(date).impute.vcf.gz - snps.Rda - A mapping snp set generated from
WI.(date).hard-filter.vcf.gz
isotype¶
This directory contains files that integrate with the genome browser on CeDNR.
isotype/tsv/¶
This directory contains tsv's that are used to show where variants are in CeNDR.
isotype/vcf/¶
This direcoty contains the VCFs of isotypes.
phenotype/¶
- MT_content.tsv - Mitochondrial content (MtDNA cov / Nuclear cov).
- kmers.tsv - 6-mers for each isotype.
- telseq.tsv - Telomere length for each isotype ~ split out by read group.
popgen/¶
- WI.(date).tajima.bed.gz - Tajima's D bedfile for use on the report viewer of CeNDR.
- WI.(date).tajima.bed.gz.tbi - Tajima's D index.
- trees/ - Phylogenetic trees for each chromosome and the entire genome.
- I.(pdf|png|tree)
- ...
- genome.(pdf|png|tree)
report/¶
- multiqc.html - A comprehensive report of the sequencing run.
- multiqc_data/
- multiqc_bcftools_stats.json
- multiqc_data.json
- multiqc_fastqc.json
- multiqc_general_stats.json
- multiqc_picard_AlignmentSummaryMetrics.json
- multiqc_picard_dups.json
- multiqc_picard_insertSize.json
- multiqc_samtools_idxstats.json
- multiqc_samtools_stats.json
- multiqc_snpeff.json
- multiqc_sources.json
track/¶
- (date).LOW.bed.gz(+.tbi) - LOW effect mutations bed track and index.
- (date).MODERATE.bed.gz(+.tbi) - MODERATE effect mutations bed track and index.
- (date).HIGH.bed.gz(+.tbi) - HIGH effect mutations bed track and index.
- (date).MODIFIER.bed.gz(+.tbi) - MODERATE effect mutations bed track and index.
- phastcons.bed.gz(+.tbi) - Phastcons bed track and index.
- phylop.bed.gz(+.tbi) - PhyloP bed track and index
Variation/¶
- WI.(date).soft-filter.vcf.gz(+.csi|+.tbi) -
- WI.(date).soft-filter.stats.txt -
- WI.(date).hard-filter.vcf.gz(+.csi|+.tbi) -
- WI.(date).hard-filter.stats.txt -
- WI.(date).hard-filter.genotypes.tsv -
- WI.(date).hard-filter.genotypes.frequency.tsv -
- WI.(date).impute.vcf.gz(+.csi|+.tbi) -
- WI.(date).impute.stats.txt -
- sitelist.tsv.gz(+.tbi) - Union of all sites identified in original SNV variant calling round.