The genetic basis of natural variation in C. elegans telomere length
Cook et al.

Cook DE, Zdraljevic S, Tanny RE, Seo B, Riccardi DD, Noble LM, Rockman MV, Alkema MJ, Braendle C, Kammenga JE, Wang J, Kruglyak L, Félix MA, Lee J, Andersen EC
(2016 Sep 1) Genetics [ DOI | Pubmed ]
Abstract
Telomeres are involved in the maintenance of chromosomes and the prevention of genome instability. Despite this central importance, significant variation in telomere length has been observed in a variety of organisms. The genetic determinants of telomere-length variation and their effects on organismal fitness are largely unexplored. Here, we describe natural variation in telomere length across the Caenorhabditis elegans species. We identify a large-effect variant that contributes to differences in telomere length. The variant alters the conserved oligosaccharide/oligonucleotide-binding fold of POT-2, a homolog of a human telomere-capping shelterin complex subunit. Mutations within this domain likely reduce the ability of POT-2 to bind telomeric DNA, thereby increasing telomere length. We find that telomere-length variation does not correlate with offspring production or longevity in C. elegans wild isolates, suggesting that naturally long telomeres play a limited role in modifying fitness phenotypes in C. elegans.
Datasets
File S01
Strain isotype grouping and collection information
File S02
Sequence run information
File S03
Rates of heterozygosity for different SNV calling methods
File S04
Summary of variant filters.tsv
File S05
Telomere Hexamer Locations Across WS245
File S06
Telseq length estimates by sequencing run
File S07
P-values from MMP enrichment test
File S08
TelSeq telomere length estimates among wild isolates
File S09
Telomere length measurements comparison
File S10
QTL Mapping Results
File S11
QTL Confidence Interval Genes
Imputed variant set
SNVs imputed with Beagle
Annotated variant set
SNVs annotated with SnpEff
Supplementary Figures
- Figure S01 - Evaluation of variant-calling parameters from a simulated variant data set
- Figure S02 - Pairwise SNV concordance among isotypes
- Figure S03 - Frequency of hexamers in short reads generated from the N2 strain
- Figure S04 - Terminal restriction fragment (TRF) Southern blot Analysis
- Figure S05 - Correcting MMP TelSeq estimates for differences in read length
- Figure S06 - Distribution of the depth of coverage among 152 isotypes
- Figure S07 - Distribution of number of variant sites as compared to the WS245 reference genome
- Figure S08 - Distribution of SNVs by chromosome
- Figure S09 - Similarity tree of 152 C. elegans isotypes
- Figure S10 - TelSeq differences by DNA fragmentation method
- Figure S11 - Lack of a significant correlation between depth of coverage and TelSeq length estimate
- Figure S12 - Longevity is not associated with telomere length
- Figure S13 - Tajima’s D does not support evidence of selection at the pot-2 locus
- Figure S14 - Similarity trees of genome, chromosome II, and the pot-2 locus
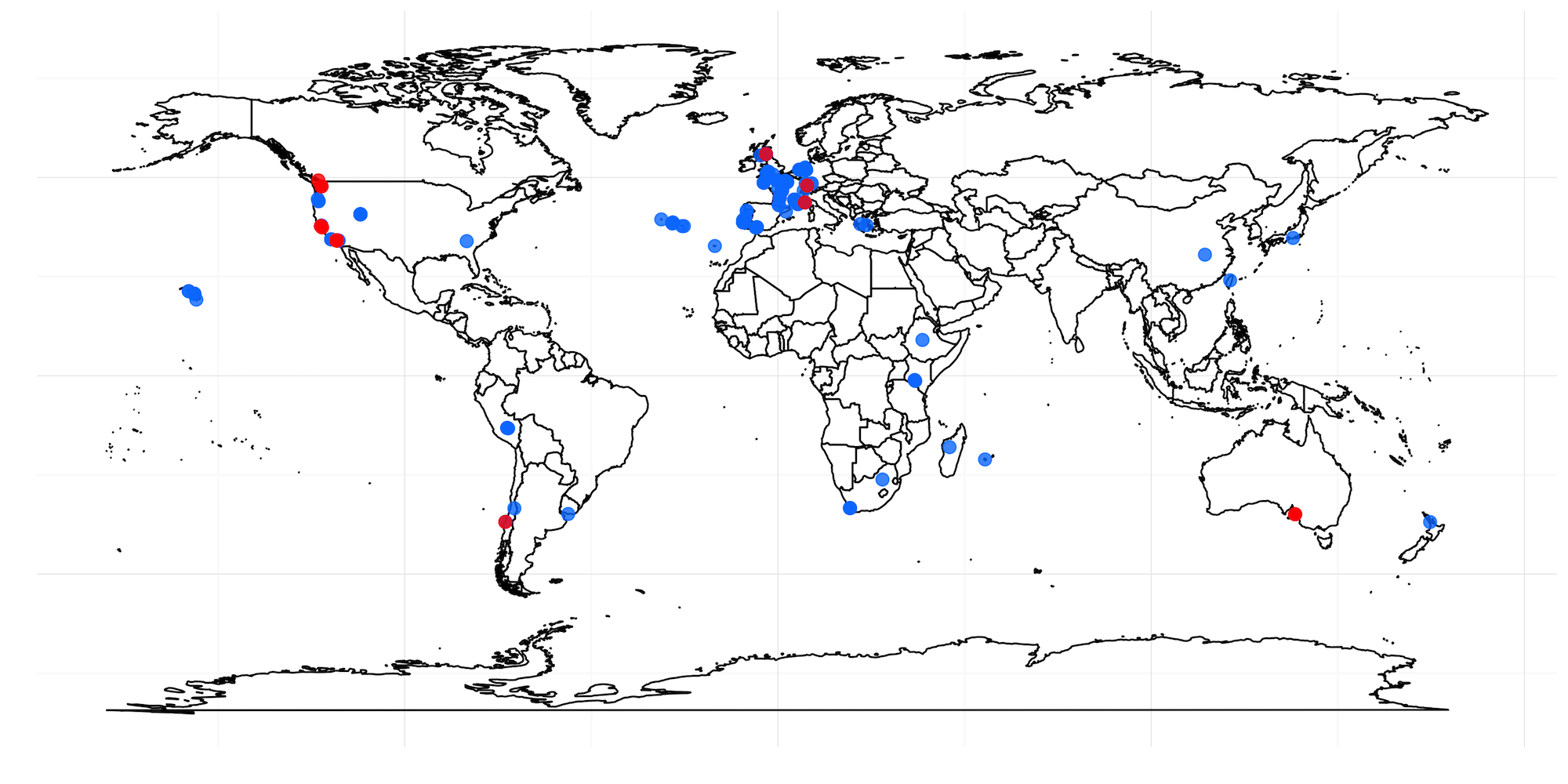
- Figure S15 - Strains with the F68I variant are not found in similar geographic locations
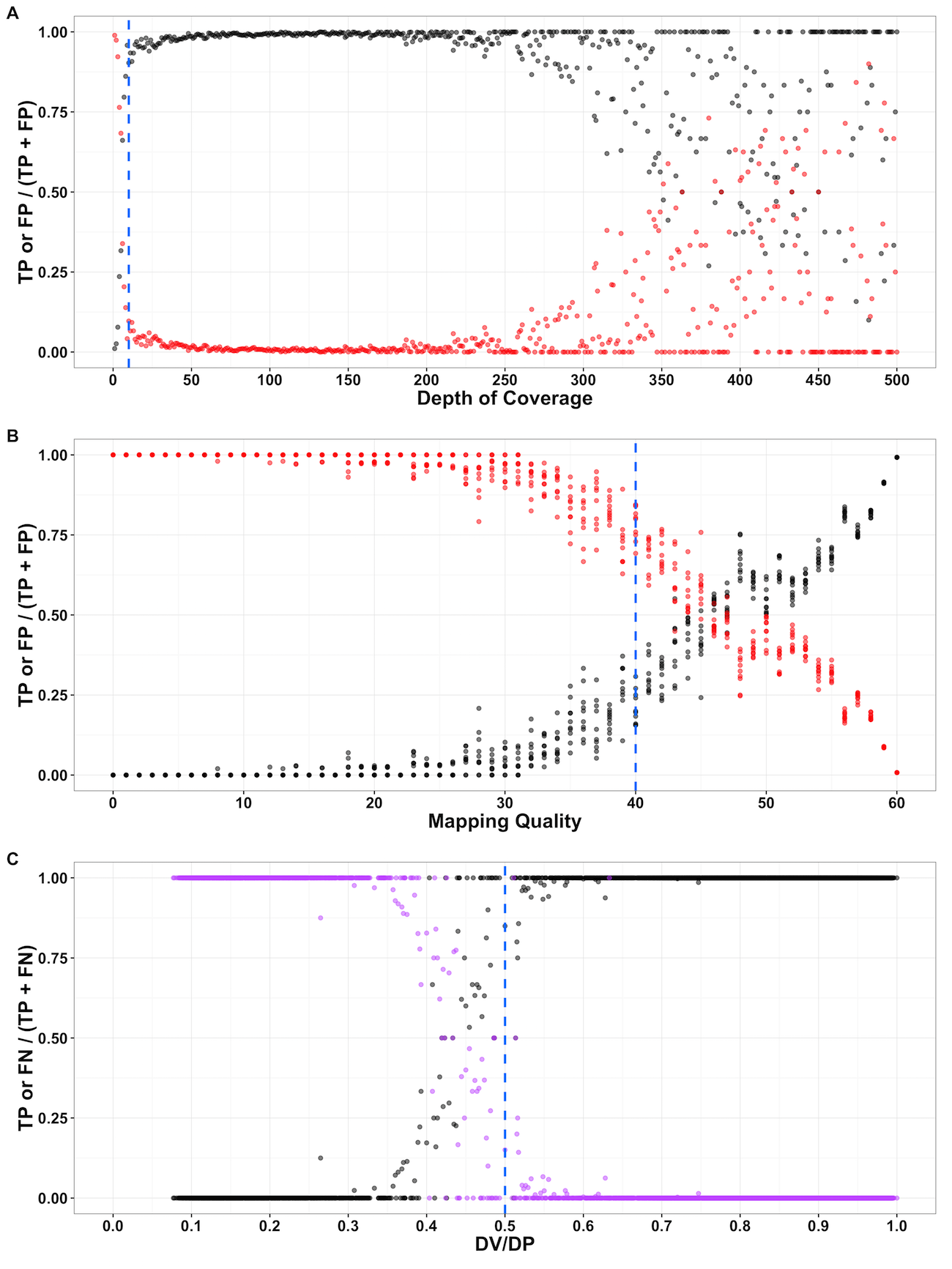
Figure S01 - Evaluation of variant-calling parameters from a simulated variant data set
True positive (TP; black), false positive (FP; red), and false negative (FN; purple) rates across variant parameters suggest appropriate filter thresholds. The y-axis is the rate is the rate of TP compared with either FP or FN variants. The rate of TP and FP or FN is plotted on the x-axis across (A) depth of coverage, (B), mapping quality, or (C) the number of high-quality non-reference bases (DV) over depth (DP) at a given site. allele for that isotype (F = Red, I = Blue).
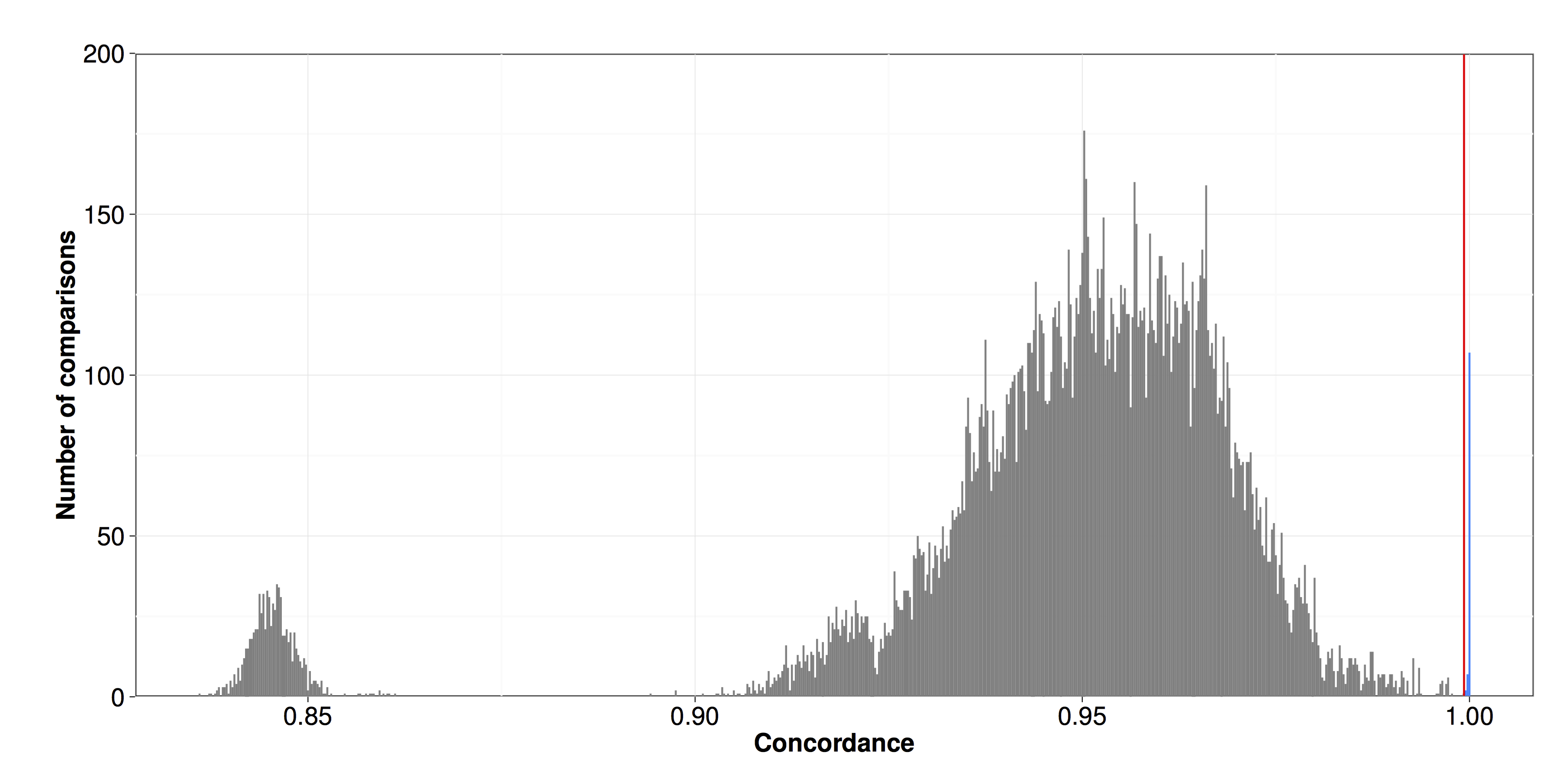
Figure S02 - Pairwise SNV concordance among isotypes
Pairwise SNV concordances among all strains were assessed by comparing SNV calls between pairs of strains. Concordance is plotted on the x-axis. The red line indicates the cutoff used to assign isotypes (0.9993). Strains with concordances above this level were classified as isotypes and are colored in blue.
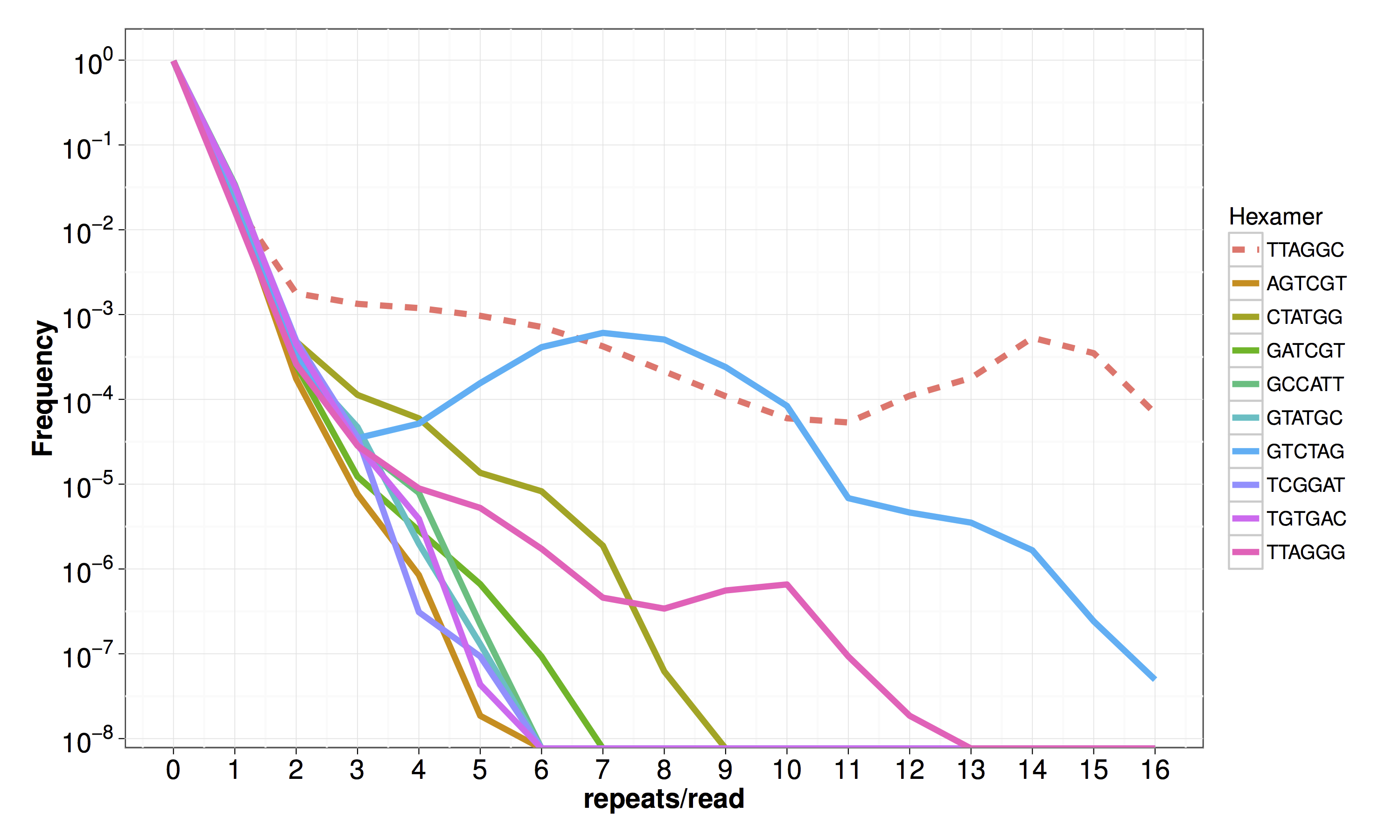
Figure S03 - Frequency of hexamers in short reads generated from the N2 strain
The frequency of non-cyclical permutations of the telomeric hexamer is plotted on the y-axis against the number of hexamer repeats per read on the x-axis. The line color indicates the hexamer. The dashed line represents the telomeric hexamer repeat in C. elegans.
 Southern blot Analysis.png)
Figure S04 - Terminal restriction fragment (TRF) Southern blot Analysis
Terminal restriction fragment analysis was performed on a subset of wild strains to evaluate the accuracy of TelSeq telomere-length estimates. Genomic DNA from wild isolates was separated by pulsed-field gel electrophoresis. Equivalent amounts of DNA were loaded into each lane.
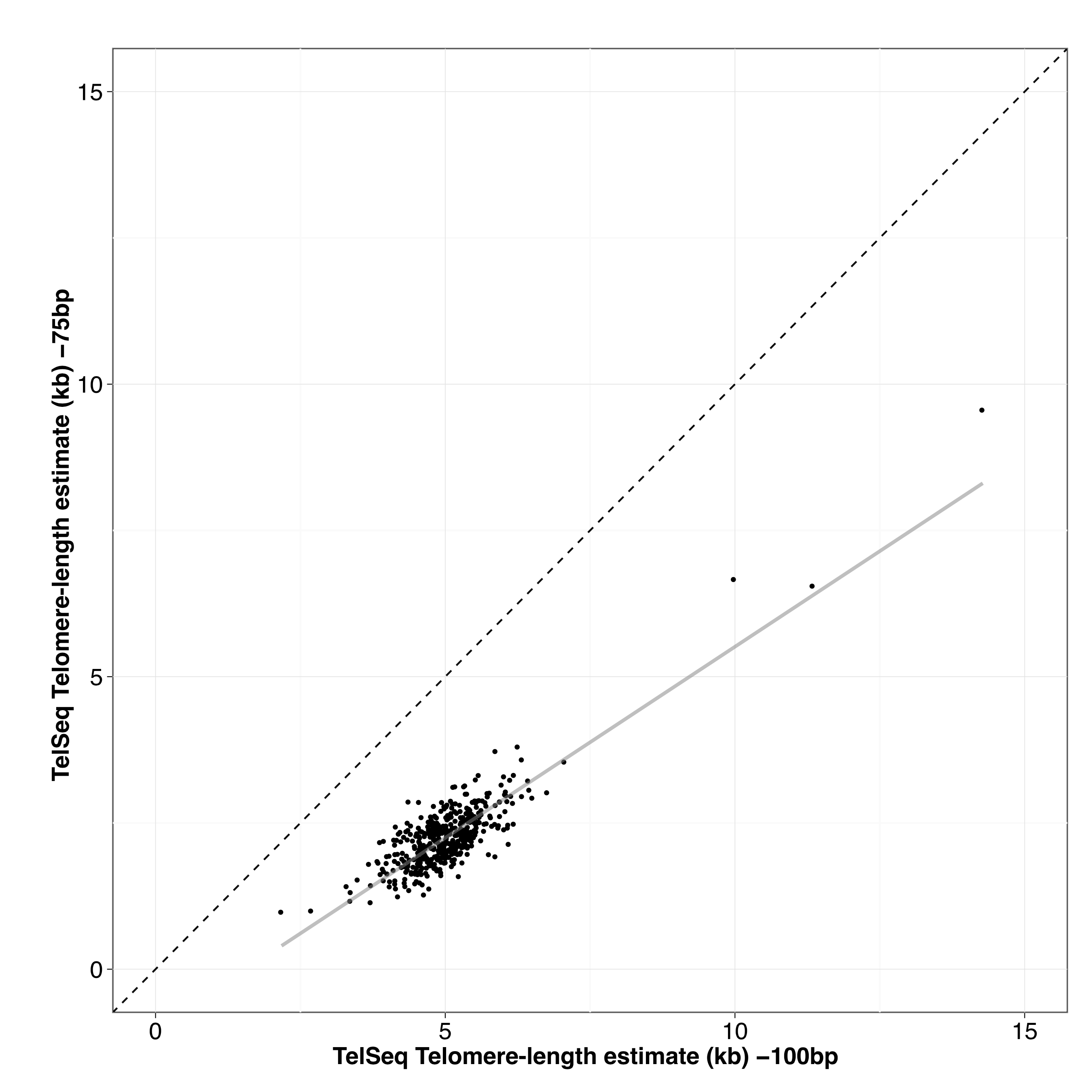
Figure S05 - Correcting MMP TelSeq estimates for differences in read length
TelSeq telomere-length estimates from 448 Million Mutation Project strains sequenced at 75 bp and 100 bp were used to develop a linear model for converting TelSeq estimates. This transformation enabled us to convert TelSeq estimates using sequence data from 75 bp runs. The correlation between 75 bp and 100 bp estimates was r2 = 0.723.
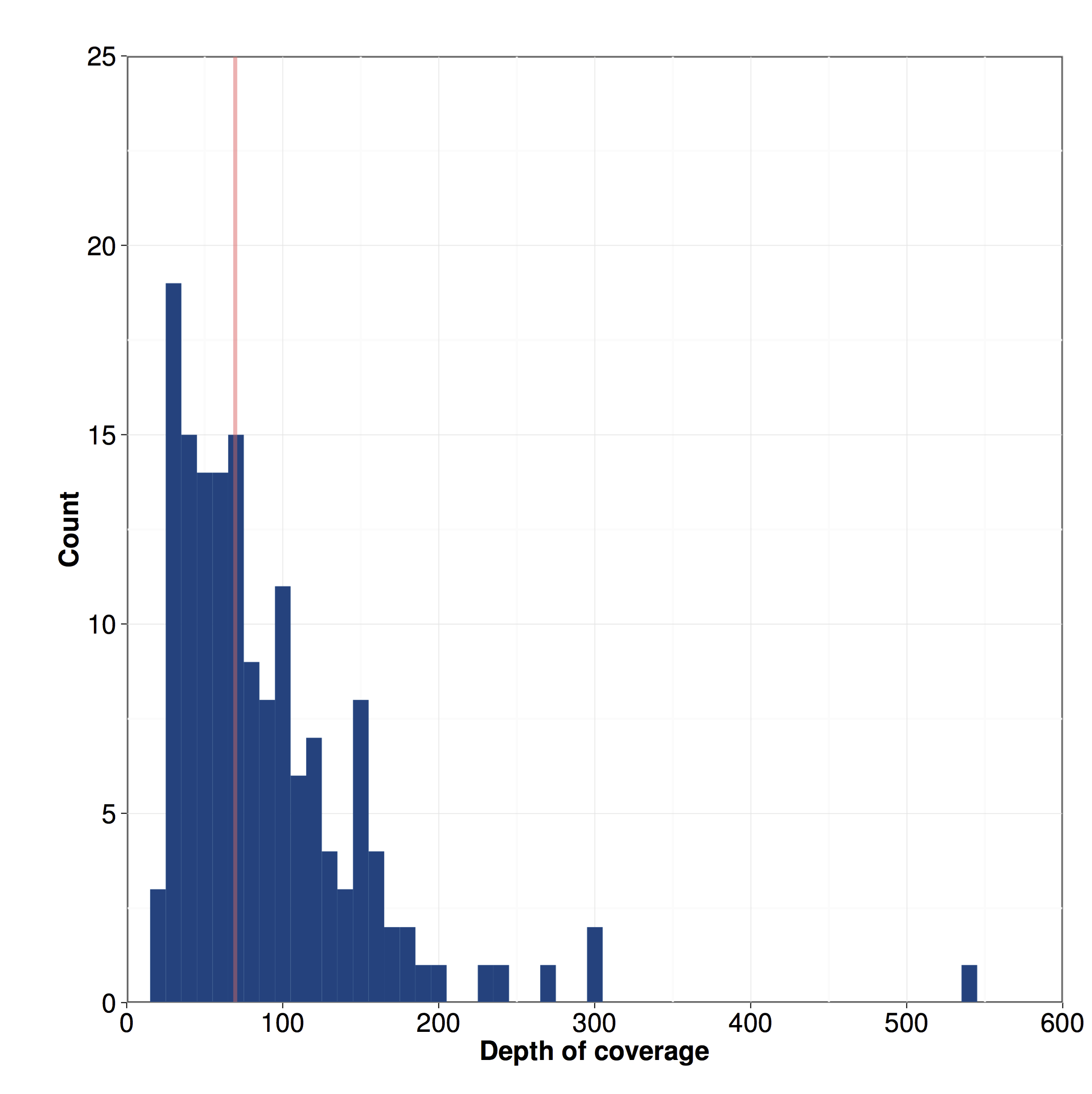
Figure S06 - Distribution of the depth of coverage among 152 isotypes
The distribution of depth of coverage for 152 isotypes is shown. The red line indicates median depth of coverage: 70x.
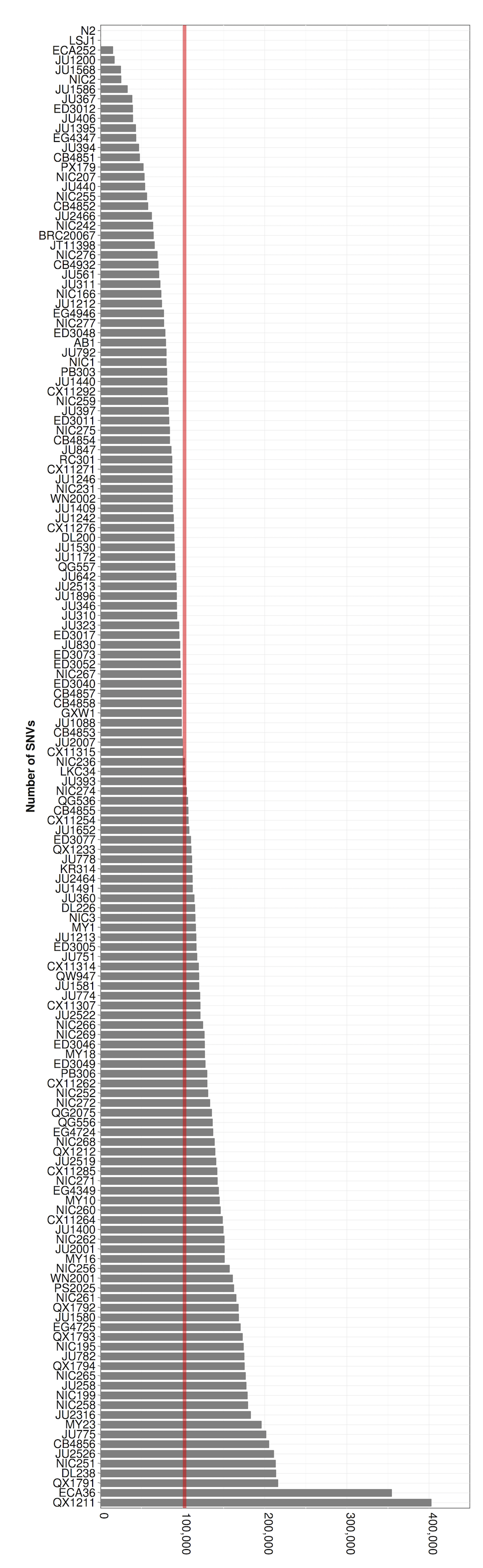
Figure S07 - Distribution of number of variant sites as compared to the WS245 reference genome
Number of single nucleotide variants (SNVs) is plotted on the x-axis for each isotype (y-axis).
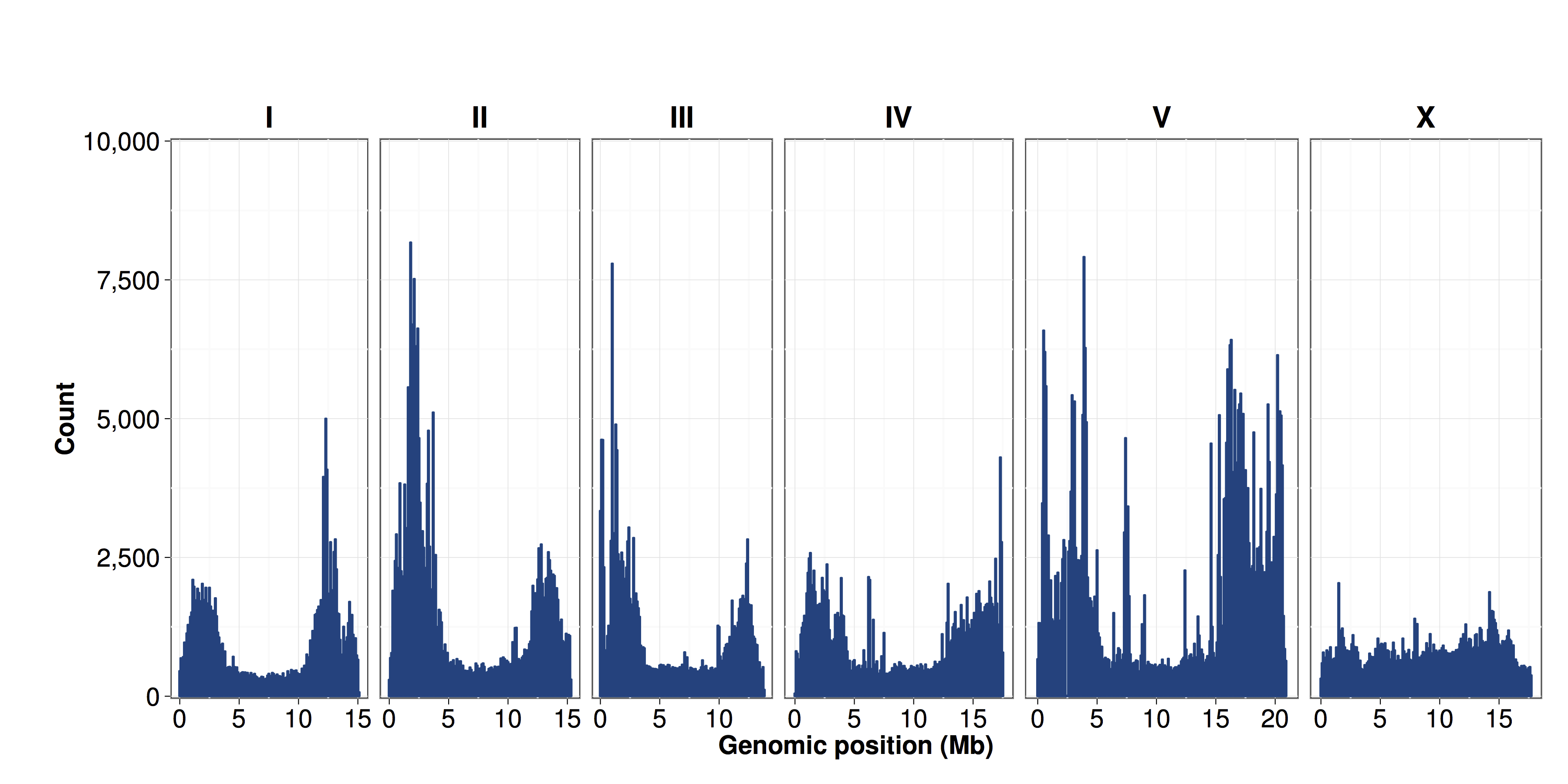
Figure S08 - Distribution of SNVs by chromosome
The number of SNV sites identified from the 152 isotypes relative to the N2 strain were calculated in 100 kb bins and plotted by genomic position (x-axis). The density of SNVs is greater on chromosome arms than in centers.

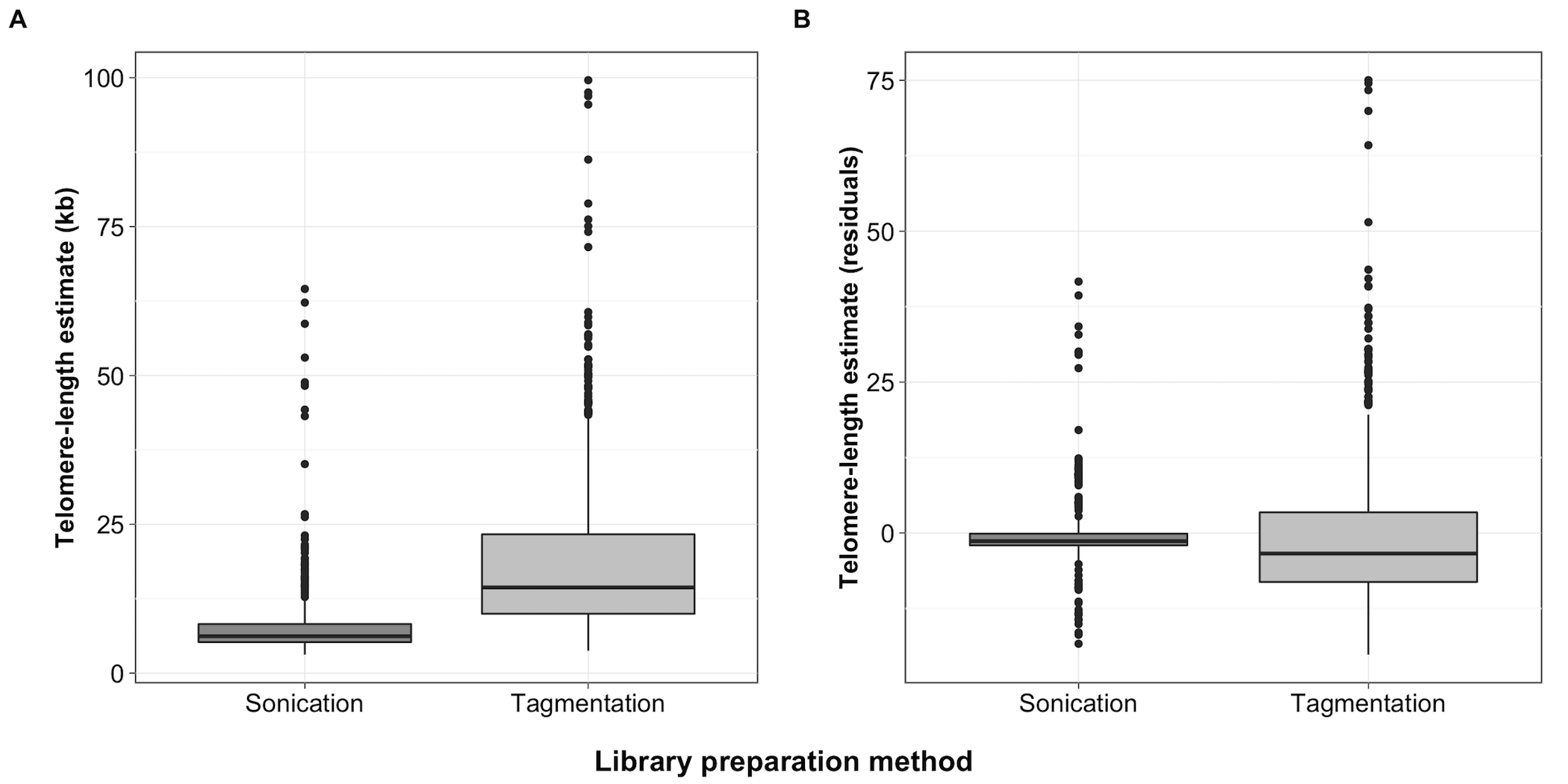
Figure S10 - TelSeq differences by DNA fragmentation method
Differences in TelSeq length estimates based on DNA fragmentation method shown as boxplots. Dark lines within the center of each box represent the median, and the box represents the interquartile range (IQR) from the 25th-75th percentile. Whiskers extend to 1.5x the IQR above and below the box. Points represent estimates beyond 1.5x the IQR. (A) Uncorrected TelSeq length estimates are plotted on the y-axis by DNA fragmentation method. (B) Residual telomere length estimates are plotted by DNA fragmentation method.
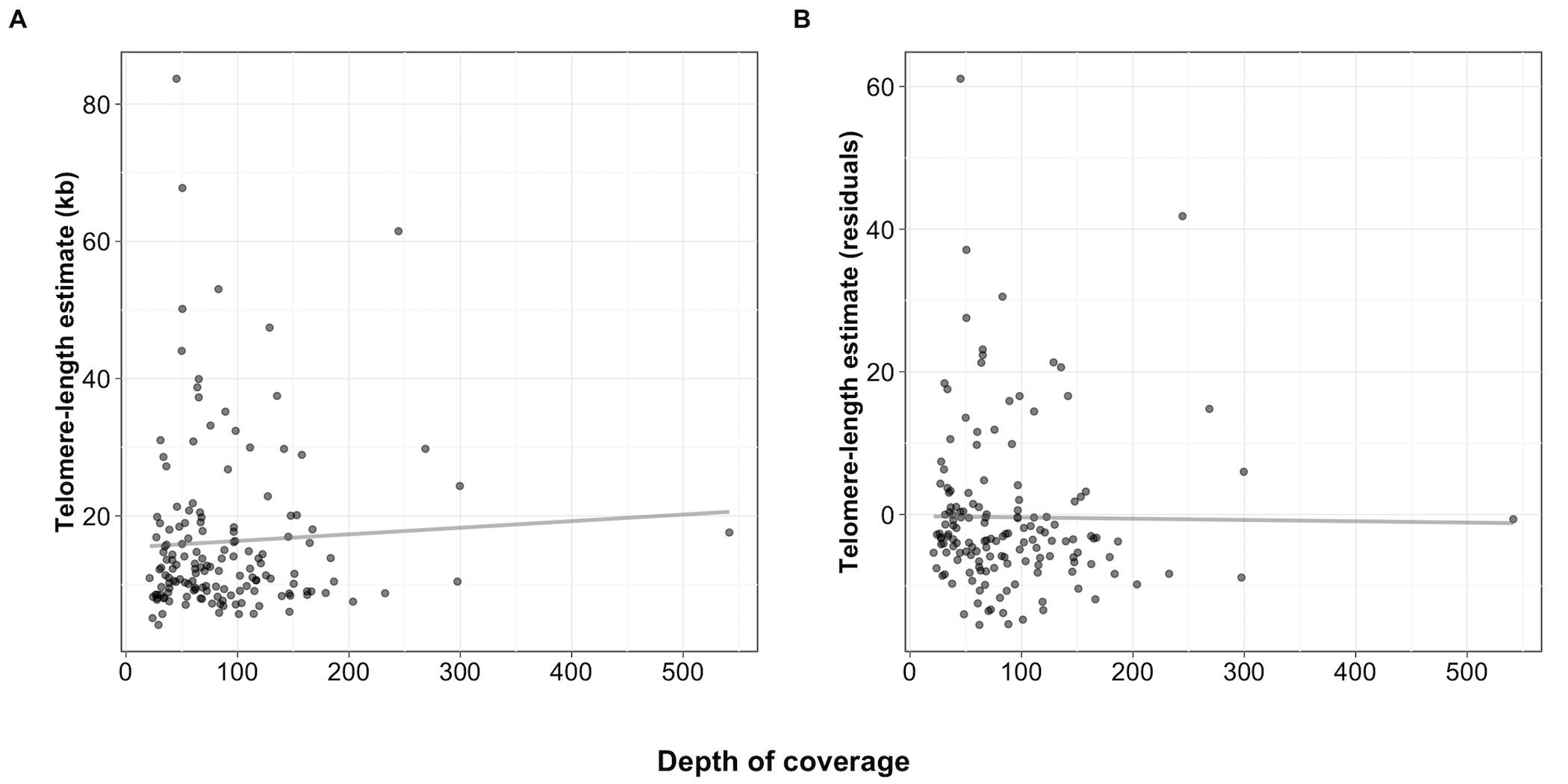
Figure S11 - Lack of a significant correlation between depth of coverage and TelSeq length estimate
Scatterplots of the depths of coverage for isotypes on the x-axis plotted against (A) the TelSeq telomere-length estimates (rho = 0.045, p = 0.576). (B) Depths of coverage are plotted against the residuals of the TelSeq length estimates after adjusting for sequencing center (run) and sequencing library (rho = -0.134, p = 0.097).
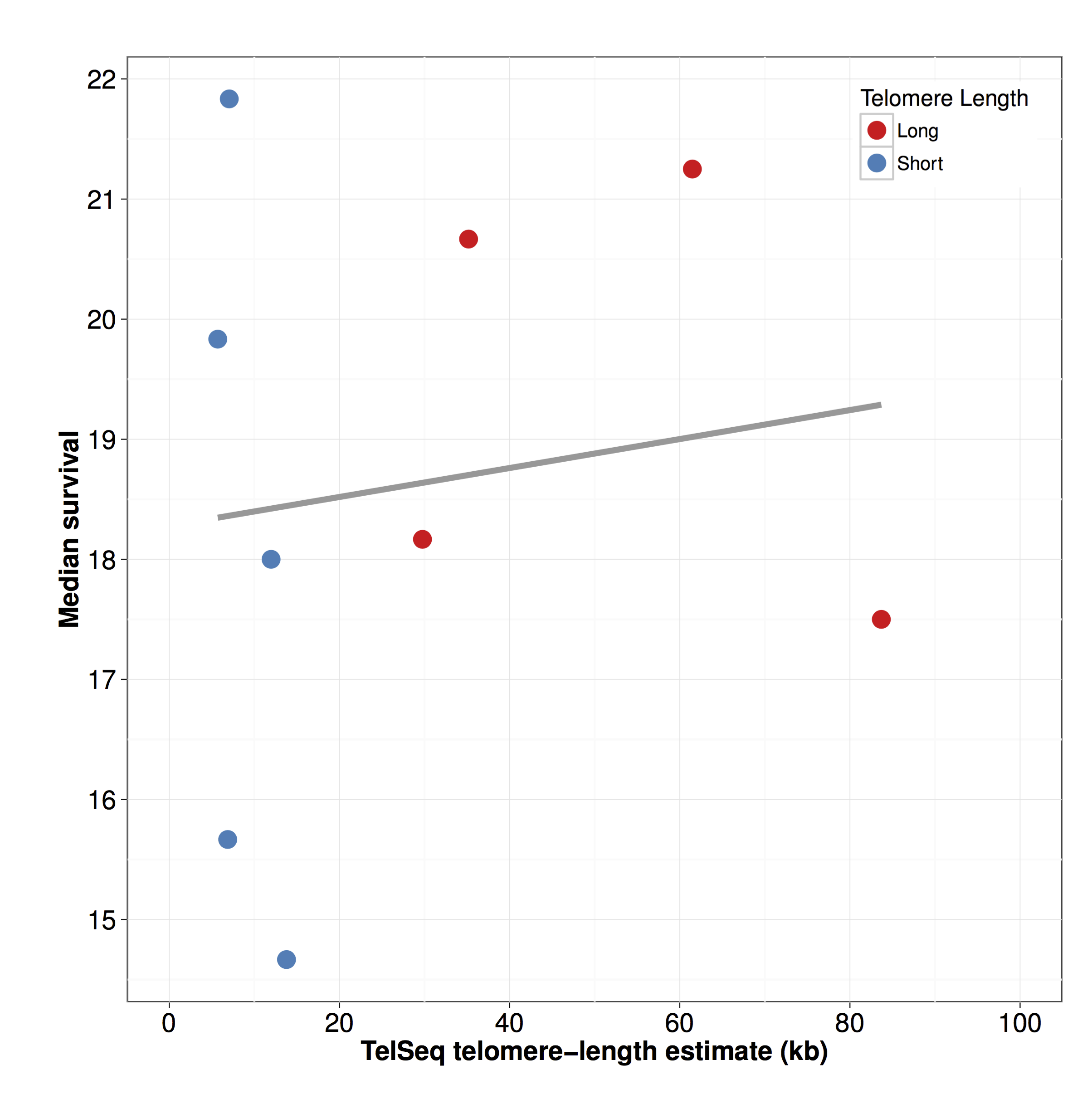
Figure S12 - Longevity is not associated with telomere length
Median day of survival (x-axis) is plotted against the estimated telomere length (y-axis). The correlation is not significant (rho = 0.05 , p = 0.91).
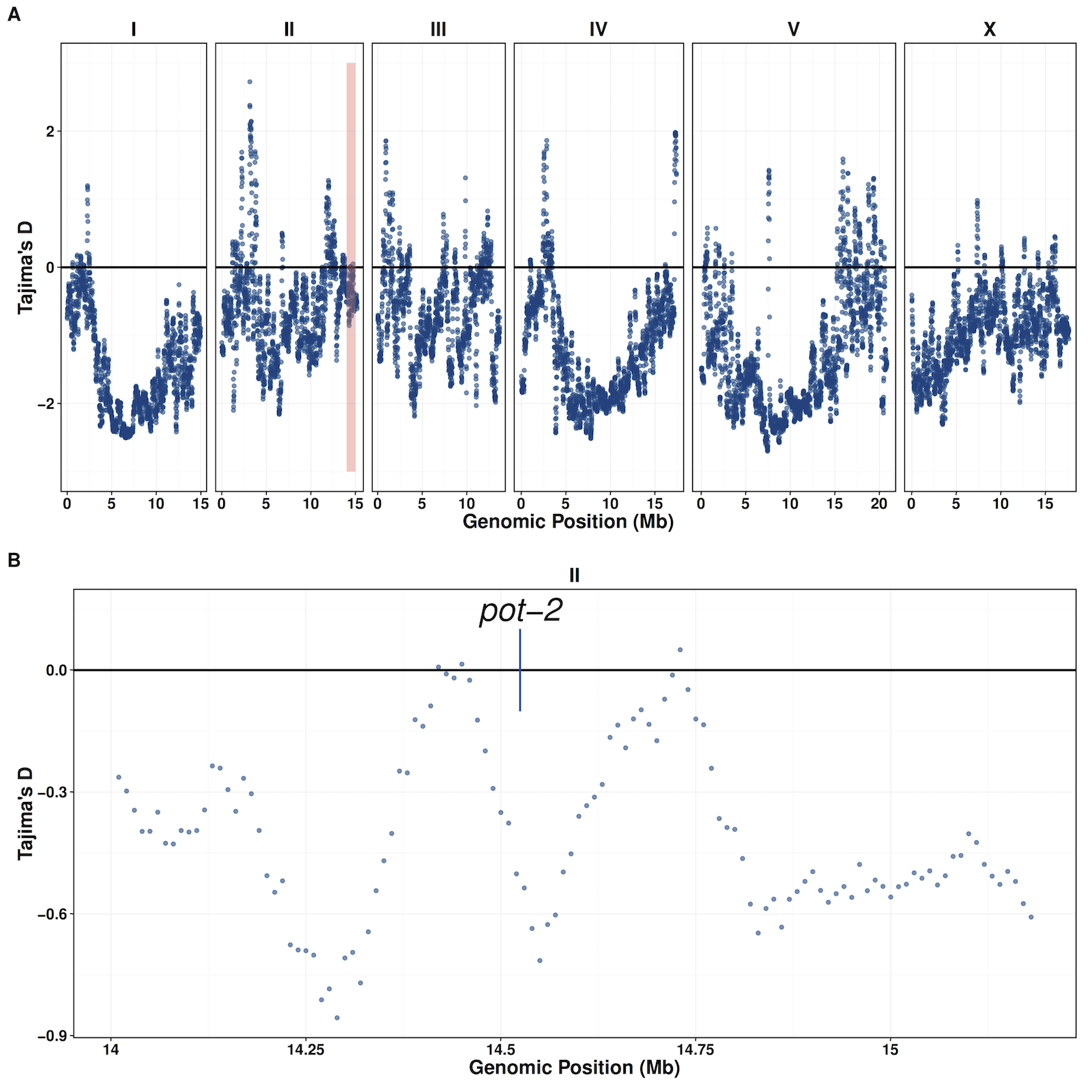
Figure S13 - Tajima’s D does not support evidence of selection at the pot-2 locus
Tajima’s D values were calculated genome wide (window-size = 100,000, step-size = 10,000). (A) Distribution of Tajima’s D values plotted by genomic position. The red box represents the region shown in panel (B) that contains the pot-2 locus.

Figure S14 - Similarity trees of genome, chromosome II, and the pot-2 locus
Isotypes are colored by their POT-2 F68I variant status. F=Blue, I=Red. Trees were generated from SNVs (A) genome-wide, (B) using SNVs located on the right arm of chromosome II (13Mb to the terminus), or (C) using SNVs located within the pot-2 region (II:14,524,173-14,525,111).
{kind=link}
{kind=link}
{kind=link}
 Southern blot Analysis.png){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}